Vasculitis leucocitoclástica fibrosante localizada crónica de manifestación insidiosa: diagnóstico sindromático
Chronic localized fibrosing leukocytoclastic vasculitis with an insidious manifestation: syndromic diagnosis.
Med Int Méx. 2020 marzo-abril;36(2):272-274. https://doi.org/10.24245/mim.v36i2.2923
León Felipe Ruiz-Arriaga,1 Erika Rodríguez-Lobato,2 Daniel Asz-Sigall,3 María Elisa Vega-Memije1
1 Departamento de Dermatopatología.
2 Departamento de Cirugía Dermatológica y Oncológica.
Hospital General Dr. Manuel Gea González, Ciudad de México, México.
3 Clínica de Oncodermatología, Universidad Nacional Autónoma de México, Ciudad de México, México.
Sr. Editor:
El propósito de esta carta es presentarle un caso de vasculitis leucocitoclástica fibrosante localizada crónica, que pertenece al espectro de las vasculitis neutrofílicas de pequeños vasos mediada por inmunocomplejos. Este tipo de vasculitis se caracteriza por su típico patrón de fibrosis, ya sea lamelar, esteriforme o concéntrico.1
La vasculitis leucocitoclástica fibrosante localizada crónica se manifiesta en diversos padecimientos, como son: el granuloma facial, el eritema elevatum diutinum, la fibrosis angiocéntrica eosinófilica, el pseudotumor inflamatorio y las enfermedades inflamatorias asociadas con IgG4.2-4
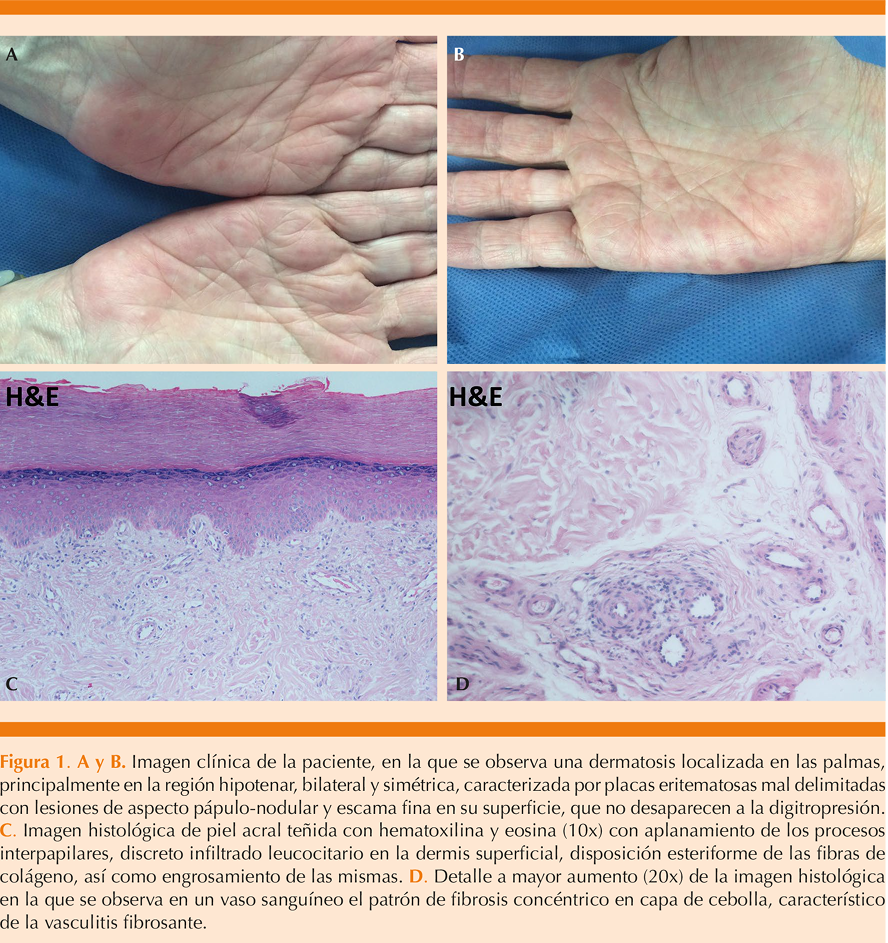
Se trata de una paciente femenina de 71 años de edad, que tenía el antecedente personal patológico de lupus eritematoso sistémico, tratada hacía 10 años con hidroxicloroquina, asintomática y actualmente sin tratamiento; quien acudió a consulta por padecer una dermatosis localizada a las palmas, bilateral, simétrica, acentuada a la región hipotenar; caracterizada por placas eritematosas, irregulares, mal definidas, que no desaparecían a la vitropresión, así como lesiones de aspecto nodular de 3 a 4 cm de diámetro (Figura 1A y B). La dermatosis tenía seis semanas de evolución y era referida como asintomática. Se realizó un estudio histopatológico con los siguientes diagnósticos diferenciales: dermatosis asociada con reactivación de lupus eritematoso sistémico y eritema multiforme.
En el estudio histológico se observó un corte de piel acral con acantosis irregular, vasos sanguíneos dilatados, discreto infiltrado inflamatorio de linfocitos y fibras de colágena engrosadas y de distribución irregular, en patrón esteriforme. Algunas fibras de colágeno perpendiculares a la epidermis en dermis papilar y otras en patrón concéntrico, en “capas de cebolla”, a vasos sanguíneos de pared engrosada e infiltrada por células inflamatorias (Figura 1C y D). La inmunotinción del tejido para IgG4 resultó negativa.
Mediante la correlación clínico-patológica se orientó el diagnóstico de esta paciente a dos afecciones:
Eritema elevatum diutinum, a favor de este diagnóstico es que este padecimiento tiene asociación con enfermedades autoinflamatorias,5 como el lupus eritematoso sistémico que padecía nuestra paciente; las lesiones dermatológicas estaban en un sitio atípico, pero ya descrito;6,7 en términos histológicos, puede corresponder a una fase crónica de la enfermedad. En contra de este diagnóstico es el hecho de que no tiene la morfología característica, que se manifiesta en placas eritemato-violáceas o marrones, de aspecto nodular o pseudoqueloideo, bien delimitadas.6-8
Enfermedad inflamatoria cutánea asociada a IgG4, a favor de este diagnóstico está la morfología de las lesiones clínicas, que pueden englobarse en la erupción inespecífica maculopapular o eritematosa de esta enfermedad;9 la topografía más habitual es en la cabeza y el cuello; no obstante, puede afectar todo el cuerpo;9 respondió favorablemente a la terapia con prednisona, lo que es un criterio mínimo de sospecha.10 Contrario a esto está que histológicamente se le pudo clasificar como histología con evidencia insuficiente para el diagnóstico según los criterios histológicos propuestos por Deshpande y colaboradores debido a que solo tenía los patrones de fibrosis característicos, pero no pudieron evidenciarse los depósitos de IgG4 en los tejidos, posiblemente debido al tratamiento previo con prednisona sistémica; de la misma manera, las concentraciones séricas de IgG4 no rebasaron los concentraciones normales. No obstante, sugerimos que a pesar de no confirmarse la enfermedad por los criterios previamente descritos, no se descarte la enfermedad.
Una vez descritas las características a favor y en contra de ambas causas, abogamos por su experiencia clínica, así como la de sus lectores, para poder clasificar y brindar un diagnóstico etiológico adecuado a esta paciente, debido a las implicaciones terapéuticas y de pronóstico de las afecciones etiológicas descritas.
REFERENCIAS
1. Carlson JA, LeBoit PE. Localized chronic fibrosing vasculitis of the skin: an inflammatory reaction that occurs in settings other than erythema elevatum diutinum and granuloma faciale. Am J Surg Pathol 1997;21(6):698-705. DOI: 10.1097/00000478-199706000-00010.
2. Deeken A, Jefferson J, Hawkinson D, Fraga G. Localized chronic fibrosing vasculitis in a tattoo: a unique adverse tattoo reaction. Am J Dermatopathol 2014;36(4):e81-e83. doi: 10.1097/DAD.0b013e3182a27a99.
3. Marcoval J, Moreno A, Peyr J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol 2004;51:269-273. DOI: 10.1016/j.jaad.2003.11.071.
4. Cesinaro AM, Lonardi S, Facchetti F. Granuloma faciale: a cutaneous lesion sharing features with IgG4-associated sclerosing diseases. Am J Surg Pathol 2013;37:66-73. doi: 10.1097/PAS.0b013e318271efd0.
5. Atallah J, Garces J, Loayza E, Carlson J. Chronic localized fibrosing leukocytoclastic vasculitis associated with lymphedema, intralymphatic and intravascular lymphocytosis, and chronic myelogenous leukemia. Am J Dermatopathol 2017;39(6):479-484. doi: 10.1097/DAD.0000000000000802.
6. Ziemer M, Koehler MJ, Weyers W. Erythema elevatum diutinum: a chronic leukocytoclastic vasculitis microscopically indistinguishable from granuloma faciale? J Cutan Pathol 2011;38:876-883. doi: 10.1111/j.1600-0560.2011.01760.x.
7. Chen G, Cheuk W, Chan J. IgG4-related sclerosing disease a critical appraisal of an evolving clinicopathologic entity. Zhonghua Bing Li Xue Za Zhi. 2010 Dec;39(12):851-68.
8. Arenas R. Vasculitis neutrofílicas. In: Arenas R, ed. by. Dermatología: atlas, diagnóstico y tratamiento. 6th ed. Ciudad de México: McGraw Hill; 2015;310-312.
9. Tokura Y, Yagi H, Yanaguchi H, Majima Y, Kasuya A, Ito T, et al. IgG4-related skin disease. Br J Dermatol 2014;171(5):959-967. DOI:10.1111/bjd.13296.
10. Deshpande V, Zen Y, Chan J, Yi E, Sato Y, Yoshino T, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol 2012;25(9):1181-1192. doi: 10.1038/modpathol.2012.72.
Sin comentarios